超高效液相色譜-串聯質譜法測定動物源性食品中20種磺胺類藥物殘留
2021-11-29 15:38:03 來源: 世界杯賽程預測 導刊
超高效液相色譜-串聯質譜法測定動物源性食品中20種磺胺類藥物殘留
邱慧珍*,黃鳳妹,何孝金,江建麗
(南平市食品藥品檢驗檢測中心,福建南平 353000)
通信作者:邱慧珍(1988—),漢族,福建南平人,本科,主管藥師。研究方向:食品與藥品檢驗技術。E-mail:369172198@qq.com。
通信作者:邱慧珍(1988—),漢族,福建南平人,本科,主管藥師。研究方向:食品與藥品檢驗技術。E-mail:369172198@qq.com。
摘 要:目的:建立超高效液相色譜-串聯質譜法同時檢測動物源性食品中磺胺類藥物殘留。方法:用含0.1%甲酸乙腈提取,均質離心,上清液減壓蒸幹,2.0 mL的20%甲醇水溶解,2.0 mL正己烷除酯,流動相0.1%甲酸水(含6%乙腈)
-甲醇進行梯度洗脫,采用電噴霧正離子多反應監測模式檢測。空白基質加標準物質配製工作曲線,外標法定量。結果:20種化合物在2.0~40 ng/mL線性關係良好,相關係數均大於0.99,在3個加標水平2.0 μg/kg、4.0 μg/kg和10.0 μg/kg上,平均回收率在70.4%~118.1%,相對標準偏差在1.5%~10.0%,定量限為2.0 μg/kg。結論:該方法高效、快速、靈敏度高,可用於動物源性食品中磺胺類藥物殘留的日常分析。
關鍵詞:質譜;磺胺;獸藥殘留;動物源性食品
磺胺類藥物是二氫葉酸合成酶抑製劑,對氨基苯磺酰胺是此類藥物產生藥效的基本結構,甲氧苄啶是磺胺類藥物的抗菌增效劑,抑製二氫葉酸還原酶,與磺胺類藥物合用,對細菌的生長起到雙重抑製作用。磺胺類藥物是應用廣泛的抗生素之一,被用於治療動物感染,因為它們價格低廉、毒性低,並且對常見細菌感染具有出色的抗菌活性,這些抗生素若不加控製地使用,以及不遵守停藥期指南,會導致抗生素耐藥性的發展。藥物的原形及其代謝產物可蓄積於動物的組織器官或可食性產品中(如蛋、奶)中,從而危及人們的健康。
目前獸藥殘留的檢測方法主要有液相色譜法、液相色譜-串聯質譜法、液相色譜-四級杆-飛行時間質譜法等,其中液相色譜-串聯質譜法對複雜樣品具有高靈敏度、高分辨率等優點,是獸藥殘留分析的重要手段[1-2]。《國家
世界杯賽程預測監督抽檢實施細則(2021年版)》中規定磺胺類藥物檢驗的指定方法有《動物源性食品中磺胺類藥物殘留檢測 液相色譜-質譜/質譜法》(GB/T 21316—2007)、農業部1077號公告-1-2008《水產品中17種磺胺類及15種喹諾酮類藥物殘留的測定 液相色譜-串聯質譜法》、農業部1025號公告-23-2008《動物源食品中磺胺類藥物殘留檢測 液相色譜-串聯質譜法》,判定依據有農業部公告第235號、GB 31650—2019。實施細則中規定動物源性食品中磺胺類項目以磺胺類(總量)計,如豬肉中磺胺類(總量)項目至少應包含磺胺甲基嘧啶(磺胺甲嘧啶)、磺胺甲惡唑(磺胺甲鯻唑)、磺胺二甲嘧啶、磺胺間二甲氧嘧啶(磺胺地索辛)、磺胺間甲氧嘧啶、磺胺喹惡啉(磺胺喹沙啉)和磺胺嘧啶,如檢出其他磺胺藥物殘留,一並計入磺胺類(總量)並判定。
本文著重探討建立超高效液相色譜-串聯質譜法測定動物源性食品中20種磺胺類(甲氧苄啶、磺胺二甲異嘧啶、磺胺醋酰、磺胺嘧啶、磺胺噻唑、磺胺吡啶、磺胺甲基嘧啶、磺胺二甲噁唑、磺胺對甲氧嘧啶、磺胺甲噻二唑、磺胺二甲基嘧啶、磺胺甲氧噠嗪、磺胺氯噠嗪、磺胺甲噁唑、磺胺鄰二甲氧嘧啶、磺胺間甲氧嘧啶、磺胺苯酰、磺胺苯吡唑、磺胺間二甲氧嘧啶及磺胺喹噁啉)藥物的檢測的方法。
1 材料和方法
1.1 儀器與設備
TSQ Quantis熱電液相質譜儀;Milli-Q超純水機(美國密理博);冷凍離心機(德國sigma);旋轉蒸發儀(德國IKA);均質器(德國IKA)。
1.2 試劑與材料
甲醇、乙腈、甲酸(均為色譜純,美國Thermo Fisher公司);20種磺胺類標準物質溶液單標(100 μg/mL),購於北京壇墨有限公司。
1.3 實驗方法
1.3.1 色譜條件
菲羅門kinetex色譜柱(100 mm×3 mm,2.6 μm);流動相:A:0.1%甲酸水(含6%乙腈),B:甲醇;梯度洗脫程序:0~6 min,25%~45%B;6~9 min,45%~85%B;9~
10 min,100% B;10~12min,100%B;12~15min,100%~25%B;流速:0.3 mL/min;柱溫:30 ℃;進樣量:10 μL。
1.3.2 質譜條件
電噴霧正離子模式(ESI+);多反應監測掃描模式(MRM);離子噴霧電壓3 500V、霧化器溫度325 ℃、傳輸管溫度325 ℃、碰撞氣N2、輔助器Ar。
1.3.3 標準工作曲線配製
將20種磺胺類標準溶液單標分別配成10 μg/mL標準貯備液,冷凍保存6個月;吸取各標準貯備液配成1.0 μg/mL混合標準溶液,冷凍保存3個月。吸取0.4 mL 1.0 μg/mL的20種磺胺類混合標準工作液於10 mL容量瓶中,乙腈定容,濃度為0.04 μg/mL(該溶液臨用現配)。稱取5份與試樣基質相應的陰性樣品2.0 g,分別加入0.04 μg/mL混合標準貯備液0.1、0.25、0.5 mL、1.0 mL、2.0 mL,按照2.6項下處理,最終得到2 ng/mL、4 ng/mL、10 ng/mL、20 ng/mL、40 ng/mL標準溶液。
1.3.4 樣品溶液的製備
準確稱取2.0 g試樣於50 mL離心管內,加入5 g無水硫酸鈉和20 mL酸化乙腈(含1%甲酸),用均質機以
10 000 r/min的速度均質30 s,振搖,將離心管置於離心機內以3500 r/min的速度離心5 min,上清液轉移至150 mL梨形瓶中,加入10 mL異丙醇,40 ℃下減壓旋轉蒸發至幹,後用2 mL的20%甲醇水溶液溶解,再加2 mL乙腈飽和正己烷渦旋洗脫,於高速離心機中15 000 r/min離心5 min分層,取下層清液用0.25 μm微孔有機濾膜過濾,上機測試。
2 結果與分析
2.1 液相方法優化與典型圖譜
水相中加入適量的甲酸,能明顯提高色譜的分離效果[3],綜合考慮色譜柱耐酸性和分離效果,添加濃度0.1%甲酸能達到滿意效果,水相中加入6%乙腈可抑製黴菌生長,減少流動相更換頻率。比較使用甲醇和乙腈作為有機相,發現使用甲醇為有機相時,色譜峰型對稱性和效果優於乙腈,最終確定0.1%甲酸水(含6%乙腈)-甲醇的組合[2]。在該色譜條件下豬肉基質陰性樣品色譜圖顯示無明顯幹擾[3],空白基質中加入磺胺類物質分離效果與質控樣圖譜疊加後如圖1所示。
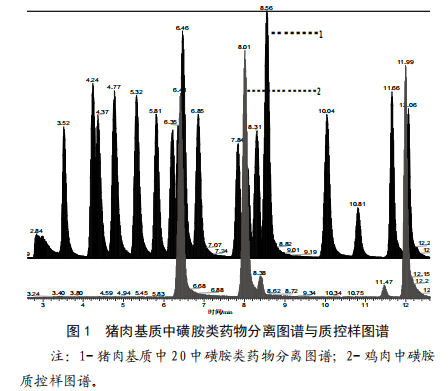
注:1-豬肉基質中20中磺胺類藥物分離圖譜;2-雞肉中磺胺質控樣圖譜。
2.2 質譜參數優化與離子信息
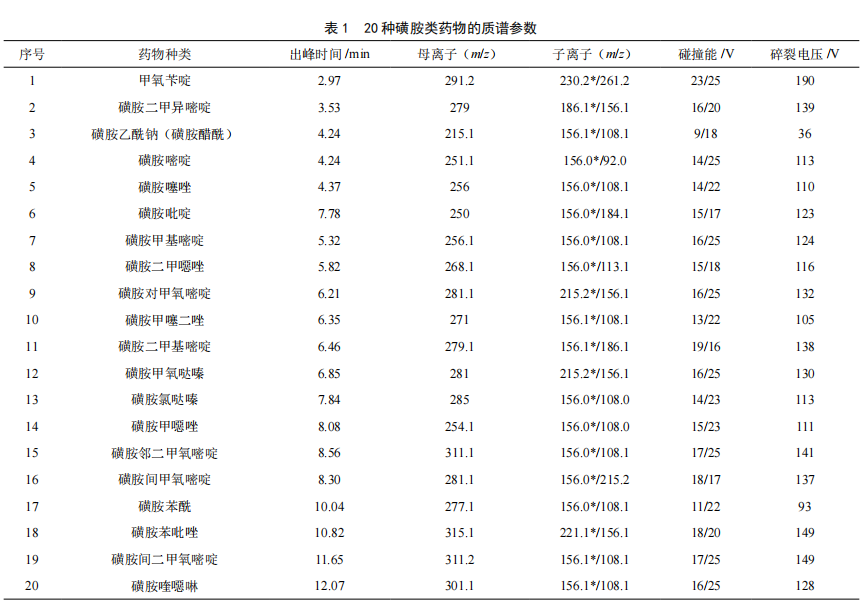
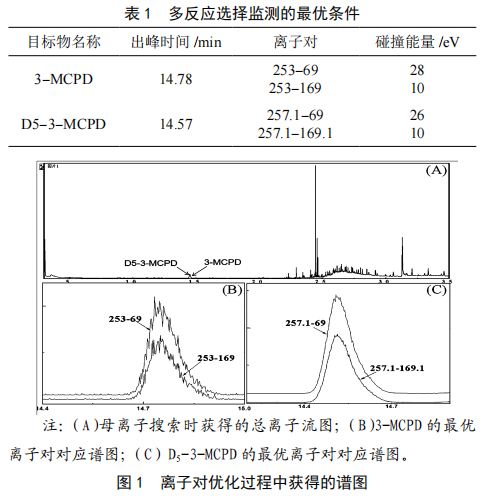
在電噴霧正離子模式(ESI+)下,分別對20種(1 μg/mL)磺胺類獸藥進行全掃(SCAN),找出母離子參數;在單離子掃描模式下(SIM)找出最佳的碎裂電壓,使母離子響應達到最大;在產生子離子模式(Product Ion)下找出一對定量和定性離子,後在多反應監測(MRM)模式下優化碎裂電壓,使子離子的響應達到最大值,優化後的參數見表1,帶*為定量離子[3-4]。
2.3 前處理方法優化
本實驗比較幾種國標方法中使用的提取液乙腈-水
(1 000+30)、乙酸乙酯、酸化乙腈(含1%甲酸),實驗發現乙腈在提取高脂肪高蛋白基質時有較好的沉澱效果[3-4];乙腈中加入適量甲酸有助於目標物與基質中蛋白分離,提高回收率[5],故選擇酸化乙腈(含1%甲酸)作為提取液。動物源性食品基質中脂肪含量普遍較高,現有淨化方法有過HLB小柱[6]、MCX小柱除脂肪或經過正己烷液液分配法除脂肪,考慮到上樣、洗脫等一係列操作煩瑣,經考察,樣品經旋蒸後加入正己烷渦旋除脂肪,離心過濾後即可得到澄清透明的液體。
2.4 溶劑效應和基質效應考查
比較分別使用純有機相和初始流動相作為上機液進行分析,用純有機相上機分析得到前沿峰,峰型較寬;使用初始流動相定容上機分析,峰型正常,對稱性好,故最後上機液應使用初始流動相定容。基質效應(Matrix Effect)是基質的存在導致目標物響應產生變化的現象。基質常常對分析過程有顯著的幹擾,並影響分析結果的準確性,可以通過同等濃度的空白基質匹配標準工作曲線與溶劑配製工作曲線之比,即離子抑製(IS)評價基質效應,離子抑製率(IS)的計算公式如下:
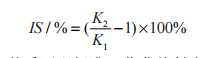
式中:K2-基質匹配標準工作曲線斜率;K1-溶劑配製標準工作曲線斜率。IS>0,基質增強;IS<0,基質抑製;IS絕對值越大基質效應越強[7-9]。
2.5 校正曲線和相關係數
取豬肉陰性基質,分別加入20種磺胺混合標液
(0.1 μg/mL)40 μL、100 μL、200 μL、400 μL、800 μL,按照1.3.4項處理得到2.0 ng/mL、5.0 ng/mL、10.0 ng/mL、20.0 ng/mL、40.0 ng/mL濃度水平。以標準溶液的保留時間為橫坐標,被測組分的峰麵積為縱坐標,繪製標準曲線。具體結果見表2。

2.6 方法的回收率、精密度
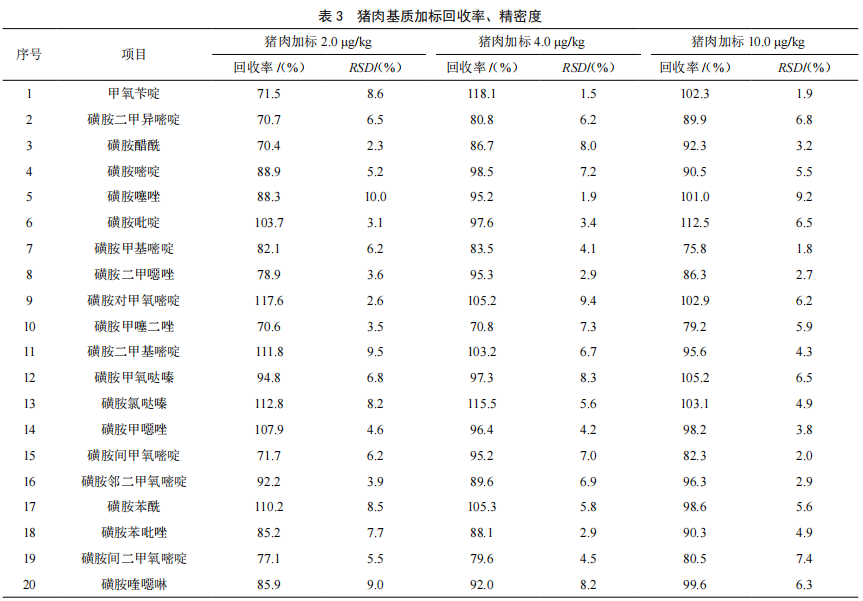
取豬肉樣品,做3個濃度水平的標準加入回收實驗,稱取樣品後分別加入20種磺胺類混合標液使樣品加標水平為2.0 μg/kg、4.0 μg/kg和10.0 μg/kg均按本方法做3次平行測定,回收率及精密度見表3。
2.7 方法的定量限
取豬肉陰性基質樣品,加標水平2.0 μg/kg,按上述方法處理,注入液相色譜-串聯質譜儀中,測定信噪比,20種磺胺類藥物均能滿足S/N≥10.0,故本方法的定量限為
2.0 μg/kg。
2.8 測定質控樣
取雞肉中磺胺質控樣(含3種磺胺類藥物)2份,按照1.3.4樣品前處理方法進行測定,測定值在標準值區間範圍內,結果如表4。

2.9 測定實際樣品
按照上述實驗條件測定監督抽檢35批雞蛋、46批禽畜肉、35批水產品,其中1批烏雞樣品磺胺類(總量)160 μg/kg(GB 31650—2019規定應≤100 μg/kg),為不合格樣品;1批雞肉樣品甲氧苄啶45 μg/kg(GB 31650—2019規定應≤
50 μg/kg),雖有檢出但未超出限量值;其餘樣品磺胺類(總量)與甲氧苄啶項目均為未檢出。
3 結論與討論
(1)農業部1077號公告-1-2008中試樣采用酸化乙腈提取,經正己烷液液萃取淨化,內標法定量。內標定量是消除基質效應的常用方法,考慮到內標物價格較貴,同時多個化合物物僅有一兩個同位素進行校正,可能造成回收不理想。同一物質在不同基質所造成的基質效應不盡相同,按樣品類別選擇相適應的空白基質可以有效消除基質效應,提高回收率。
(2)本文建立了超高效液相色譜-串聯質譜(UPLC-MS/MS)測定動物源性食品中20種磺胺類藥物殘留方法,樣品經過酸化乙腈均質提取,無水硫酸鈉除水,正己烷除油脂後上機分析,回收率在70%~120%,加標回收的RSD在10.0%。同時采用基質配工作曲線,以扣除基質效應帶來的損失。本方法與國標及其他現有方法相比,無需內標物,省去過柱等一係列煩瑣操作,具有準確、便捷、高效、靈敏的特點,可用於動物源性食品中磺胺類藥物殘留的日常分析。
參考文獻
[1]梁飛燕,盧日剛.動物源性食品中多獸藥殘留檢測方法的研究進展[J].安徽農業科學,2016,44(26):50-51.
[2]陳文俊.關於對動物源性食品中獸藥殘留檢測方法研究[J].現代食品,2019(16):129-130.
[3]陳鑫.超高效液相色譜-串聯質譜法測定動物源性食品16種獸藥殘留量的研究[J].福建輕紡,2021(3):2-9.
[4]沈春華.高效液相色譜-串聯質譜法檢測肉類原料中的藥物多殘留[J].肉類工業,2020(12):38-43.
[5]王京,王慶齡,葉佳明,等.超高效液相色譜-串聯質譜法同時測定動物源食品中30種獸藥殘留[J].分析試驗室,2016,35(8):955-960.
[6]郭添榮,柯歡,吳文林,等.動物源食品中獸藥殘留檢測方法的影響因素分析[J].農產品加工,2020(8):63-68.
[7]李紅麗.QuEChERS-UPLC-MS/MS同時測定動物性食品中24種殘留獸藥方法及基質效應的研究[D].重慶:西南大學,2020.
[8]張林田,陸奕娜,黃學泓,等.通過式淨化-高效液相色譜-串聯質譜法測定動物源性食品中42種獸藥殘留[J].分析科學學報,2020,36(1):81-87.
[9]陳剛,鄧曉軍,孫錦蘭,等.高效液相色譜-串聯質譜法同時測定動物源性食品中35種獸藥殘留量[J].理化檢驗(化學分冊),2014(7):809-814.





熱點推薦
- 2022世界杯32强赛程表时间
- 2022世界杯预选赛
-

全球食品創新平台第五期已啟動,攜手共創安全、健康、綠色的未來
-
特醫食品不是藥,卻是臨床治療的關鍵支撐
- 世界杯2022赛程时间表最新
-
超高效液相色譜-串聯質譜法測定動物源性食品中20種磺胺類藥物殘留
-
2020年上海市食用農產品監督抽檢情況分析
-
GC-MS檢測白酒中氨基甲酸乙酯方法的優化
-
存儲條件對香椿中亞硝酸鹽含量檢測的影響
-
白酒中甲醇含量的便攜式儀器快速檢測法
-
超高效液相色譜測定飲料中的食品添加劑
-
高效液相色譜-質譜法檢測水產品中硝基呋喃及其代謝物的研究
-
普洱茶熟茶(散茶)感官品質與內質分析
-

基於QuEChERs-GC-MS/MS法同時測定茶青中38種農藥殘留
-
三重四級杆氣質聯用儀法測定醬油中3-氯-1,2-丙二醇
-
微波消解-原子熒光光譜法測定食品中錫元素的方法研究
-
食用碘鹽對預防碘缺乏病的效果分析
-
基質固相分散-超高效液相色譜-串聯質譜法同時測定雞蛋中氟蟲腈
-

QuEChERS-超高效液相色譜-串聯質譜法測定雞蛋中的氯黴素類獸藥殘留
-
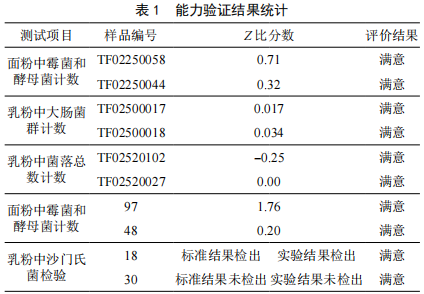
食品微生物檢驗實驗室質量控製——以陝西嘉禾藥業有限公司為例
-
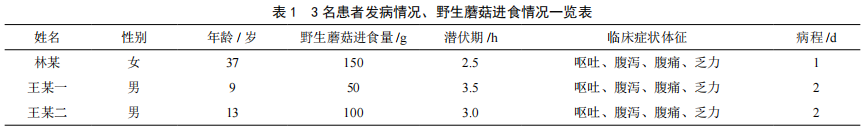
安溪縣一起青褶傘中毒事件的調查
-

哈市市場監管局執法“巡回講堂”走進哈新區
-

利用色譜方法鑒定葡萄酒中摻假組分
-
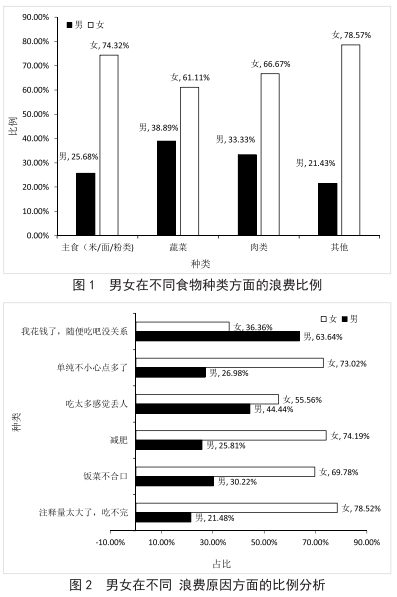
高校反食品浪費方式的探析——以廣州新華學院為例
-

食品農藥殘留檢測中樣品前處理技術探究
-

烘烤對麵包質量的影響
-

杏仁胡蘿卜酥性餅幹的工藝研究
-
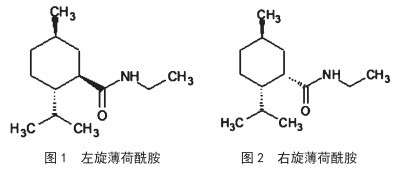
左旋薄荷酰胺的提純工藝研究
-

濕法消解-原子熒光法測定海產品總砷方法的改進
-

木瓜蛋白酶處理對牛肉品質影響的研究