三重四級杆氣質聯用儀法測定醬油中3-氯-1,2-丙二醇
2021-11-29 14:37:58 來源: 世界杯賽程預測 導刊
三重四級杆氣質聯用儀法測定醬油中3-氯-1,2-丙二醇
畢 軍
(盤錦檢驗檢測中心,遼寧盤錦 124000)
摘 要:本文建立了一種使用三重四級杆氣質聯用儀測定醬油中3-氯-1,2-丙二醇的方法。本方法使用D5-3-MCPD作為內標物,將醬油試樣經固相萃取柱淨化後,利用七氟丁酰基咪唑進行衍生,采用三重四級杆氣質聯用儀測定。結果表明:該方法在0.005~1.600 µg/mL,線性關係良好,r=0.999 3,LOD為0.002 mg/kg,LOQ為0.005 mg/kg,回收率範圍為82%~117%,相對標準偏差範圍為0.7%~7.1%。本方法定性定量準確,能夠滿足醬油中3-氯-1,2-丙二醇的檢測需求。
關鍵詞:三重四級杆;醬油;3-氯-1,2-丙二醇
3-氯-1,2-丙二醇(3-MCPD)是國際公認的食品汙染物。其主要汙染來源為酸水解植物蛋白液,由殘留的甘油三酯或甘油氯化產生[1]。WHO/FAO食品添加劑聯合專家委員會曾指出,以目前的生產工藝條件下產出的醬油被攝入後,可能造成健康危害。我國也因此出台了相應檢測標準和標準限量值[2]。但是,由於醬油基質複雜、淨化困難,因此,衍生液上機後往往會出現大量雜峰,這為3-MCPD的定性定量造成了極大的困難。而質譜技術具有靈敏度高、選擇性強等特點[3-5],能夠有效應對複雜基質所帶來的檢測困難。
基於以上技術背景,本文采用固相萃取技術結合三重四級杆氣質聯對醬油中3-MCPD進行檢測。結果表明,所建立方法高效、準確,滿足檢測需求。
1 試驗部分
1.1 儀器設備和試劑耗材
1.1.1 主要試劑耗材
乙酸乙酯、正己烷、氯化鈉、無水硫酸鈉、七氟丁酰基咪唑(純度≥98%)、矽藻土小柱(填料量5 g)。
1.1.2 主要儀器設備
Thermo TSQ8000Evo三重四級杆氣質聯用儀、BSA124S型電子天平、超聲波振蕩器、氮吹儀、旋渦振蕩器、離心機、恒溫加熱箱、氣密針。
1.2 儀器測試條件
色譜柱:DB-5MS,30 m×0.25 mm×0.25 µm;色譜柱溫度:初溫50 ℃保持1.0 min,2 ℃/min升至90 ℃,40 ℃/min升至270 ℃保持5 min,10 ℃/min升至200 ℃保持2 min;進樣口溫度:280 ℃;GC-MS接口溫度:270 ℃;進樣量:1 µL;進樣方式:不分流進樣,1.5 min後開閥;載氣:氦氣;流速1.2 mL/min;電離方式:EI;離子源溫度:300 ℃。
1.3 試驗方法
1.3.1 標準曲線的配製
采用乙酸乙酯作為溶劑,分別配製3-MCPD和D5-3-MCPD標準儲備液,其濃度均為1 000 mg/L。使用正己烷作為溶劑對儲備液進行稀釋,獲得內標物(D5-3-MCPD)標準工作液和3-MCPD標準工作液,其濃度均為10 mg/L。
分別吸取3-MCPD標準工作液0.001 mL、0.005 mL、0.010 mL、0.020 mL、0.040 mL、0.080 mL、0.160 mL和
0.320 mL置於密閉具塞玻璃管中,逐一加入D5-3-MCPD標準工作液20 µL,加入正己烷至2 mL,此時獲得濃度分別為0.005 µg/mL、0.025 µg/mL、0.050 µg/mL、0.100 µg/mL、
0.200 µg/mL、0.400 µg/mL、0.800 µg/mL和1.600 µg/mL的3-MCPD標準曲線工作溶液,臨用現配。
1.3.2 樣品的提取和淨化
準確稱取醬油試樣2 g(精確至0.001 g)置於15 mL離心管中,加入D5-3-MCPD標準工作液20 µL,旋渦混勻
5 min,待淨化。
將上述混合液全部轉移至矽藻土小柱中,平衡15 min,加入正己烷淋洗兩次,每次10 mL,棄去流出液。用15 mL乙酸乙酯對固相萃取柱上的氯丙醇進行洗脫,收集洗脫液於15 mL玻璃離心管內。使用氮氣將上述洗脫液吹至近幹(約0.1 mL,切記不可完全吹幹),用2 mL正己烷溶解上述液體獲得淨化液,將其轉移至密閉性較好的15 mL透明具塞玻璃管(閉蓋後,可用封口膜再次密封,切記不可漏氣影響衍生)中,待衍生化。
1.3.3 衍生化
用氣密針向淨化液和係列標準工作液中逐一加入40 µL七氟丁酰基咪唑。然後,立即閉蓋密封。旋渦混合30 s後,置於恒溫加熱箱內進行衍生,溫度保持為70 ℃,衍生時長為20 min。衍生後,取出冷卻至室溫,加入2 mL飽和氯化鈉溶液(或2~3 g氯化鈉),渦旋混合1 min,使水相和正己烷完全分層,且水相澄清。將正己烷盡量多的吸出,置於預先加入約0.3 g無水硫酸鈉離心管中進行幹燥,幹燥後的正己烷溶液轉移至進樣小瓶中,待質譜上機測定。
2 結果與分析
2.1 質譜條件的優化
本試驗利用TraceFinder軟件的全自動SRM離子對優化功能(AUTO SRM),對3-MCPD和D5-3-MCPD在三重四級杆質譜聯用儀中的多反應選擇監測條件進行優化。以獲得最優的質譜測定條件。
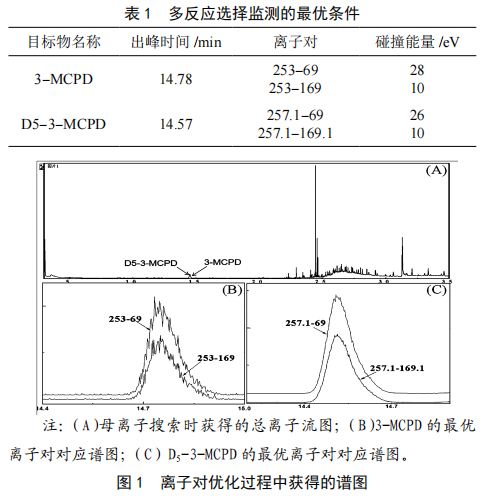
①取2 mL3-MCPD標準工作液加入D5-3-MCPD標準工作液20 µL,按照“1.3.1~1.3.3”進行操作,衍生化後上機測定。在全掃描分析模式(母離子搜索)下,測得總離子流圖,如圖1(A)所示。經分析可知,3-MCPD的出峰時間為14.78 min,響應信號最強的離子碎片為253 m/z,D5-3-MCPD的出峰時間為14.57 min,響應信號最強的離子碎片為253 m/z;②選擇子離子搜索模式,以響應信號最強的離子碎片作為母離子,在選擇離子監測分析模式下對3-MCPD和D5-3-MCPD的子離子選擇進行優化,經多個離子對進行比較可得,3-MCPD的最優離子對為253-69(定量)和253-169(定性),D5-3-MCPD的最優離子對為257.1-69(定量)和257.1-169(定性),上述兩對離子對應的譜圖如圖1(B)、圖1(C)所示;③再次進樣,對碰撞能量進行優化。最終獲得3-MCPD和D5-3-MCPD的最優多反應選擇監測條件,結果如表1所示。
2.2 淨化方式的選擇
現階段為了方便操作,醬油中氯丙醇的提取、淨化一般采用成品的矽藻土固相萃取柱。而此步驟除了可以采用直接購買的固相萃取柱外,也可以使用矽藻土填料手動裝填玻璃層析柱的方式進行替代,具體方法為將一部分填料潤濕後加入到預先裝有玻璃棉的層析柱內,再將試樣與吸附填料充分混合後,裝填至上述吸附柱內,吸附柱頂端加入一定量的無水硫酸鈉,最後進行清洗、洗脫。比較兩種方法,前者更加方便,適合大批量檢測,並且試驗的平行性較好;而後者則由於需要將試樣和填料混合均勻後再進行裝填、淨化,這使得目標物的吸附效果更佳,在不考慮裝填手法的情況下,淨化效果會略有提升。
2.3 線性範圍、檢出限和定量限
本次試驗配製了濃度為0.005~1.600 µg/mL範圍內的3-MCPD標準曲線工作溶液,其內標物(D5-3-MCPD)標準工作液的加入量為20 µL。將上述溶液按照“1.3.3”中規定進行衍生化,之後上機測定。以工作溶液中3-MCPD的濃度為橫坐標(x),以儀器軟件按內標法計算得到的衍生物峰麵積為縱坐標(y),繪製標準曲線(y=a+bx),標準曲線信息如表2所示。經分析可知,3-MCPD在給定的標準曲線範圍內,呈現出良好的線性關係。以標準曲線中間濃度水平添加空白樣品,經衍生後上機測定,之後將上述溶液使用正己烷進行逐級稀釋,並逐一測定,分別計算每個濃度水平下目標物的峰高與周圍噪聲峰高的比值(即信噪比)。試驗過程中以3倍信噪比作為方法的檢出限(LOD),以10倍信噪比作為方法的定量限(LOD),經試驗比較可知,本試驗方法中3-MCPD的LOD為0.002 mg/kg,LOD為0.005 mg/kg。
2.4 準確度和精密度
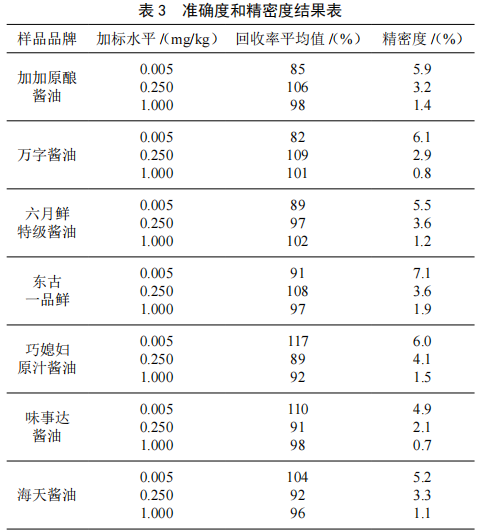
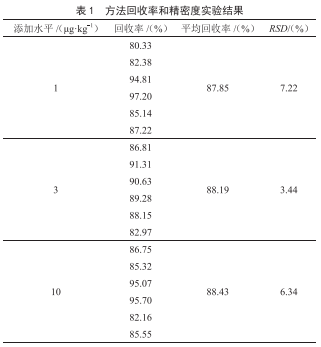
選用7種品牌的醬油作為空白基質,選取3個加標水平分別為LOD、50LOD和200LOD,全部加標試樣,按照“1.3.1~1.3.3”中規定進行前處理,按照“2.1”中最優質譜條件進行上機測定,每個濃度水平重複測定6次,通過高、中、低3個濃度點的加標回收試驗來對本文所建立方法的準確度和精密度進行評價。結果如表3所示。經測定分析得,在3個加標濃度水平下,3-MCPD在空白基質中的回收率範圍為82%~117%,精密度(RSD)範圍為0.7%~7.1%。上述結果表明該方法具有良好的準確度和精密度。
3 結論
本次研究建立一種利用三重四級杆氣質聯儀測定醬油中3-MCPD的方法。試驗中采用D5-3-MCPD作為內標物,將醬油試樣經固相萃取柱淨化後,使用七氟丁酰基咪唑進行衍生後,利用優化後的質譜條件進行測定。試驗結果表明,該方法在0.005~1.600 µg/mL,線性關係良好,選擇性優異。同時,具有較高的靈敏度、準確度和精密度,為醬油中3-MCPD的日常檢測提供了技術保障。
參考文獻
[1]李祥,楊百勤,丁紅梅.水解蛋白質調味液中氯丙醇的形成及其控製[J].中國釀造,2003(3):1-4.
[2]國家衛生和計劃生育委員會,國家食品藥品監督管理總局.
世界杯賽程預測國家標準 食品中汙染物限量:GB 2762—2017[S].北京:中國標準出版社,2017.
[3]朱炳祺,金紹強,田春霞,等.多壁碳納米管分散固相萃取結合在線GPC/GC-MS/MS 技術同時檢測茶葉中40種有機磷農藥[J].分析測試學報,2018,37(4):404—410.
[4]陳曉水,邊照陽,唐綱嶺,等.氣相色譜-串聯質譜技術分析煙草中的132種農藥殘留[J].色譜,2012,30(10):1043-1055.
[5]陳增鑫,潘芸芸,閆澤文,等.氣相色譜-質譜聯用技術在乳製品檢測中的應用[J].中國乳業,2021(3):69-72.





熱點推薦
- 2022世界杯32强赛程表时间
- 2022世界杯预选赛
-

全球食品創新平台第五期已啟動,攜手共創安全、健康、綠色的未來
-
特醫食品不是藥,卻是臨床治療的關鍵支撐
- 世界杯2022赛程时间表最新
-
三重四級杆氣質聯用儀法測定醬油中3-氯-1,2-丙二醇
-
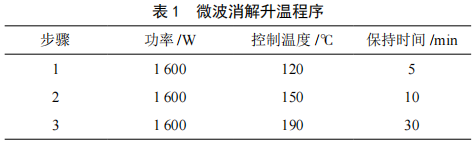
微波消解-原子熒光光譜法測定食品中錫元素的方法研究
-
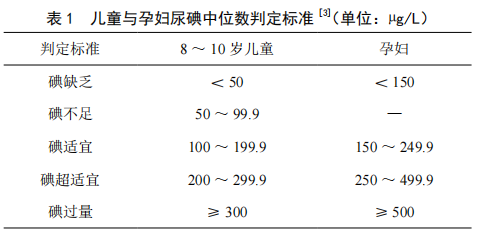
食用碘鹽對預防碘缺乏病的效果分析
-
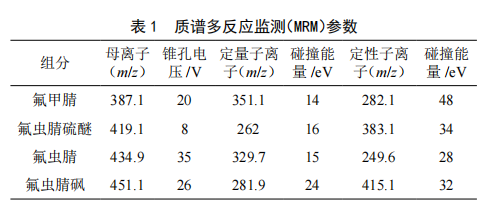
基質固相分散-超高效液相色譜-串聯質譜法同時測定雞蛋中氟蟲腈
-

QuEChERS-超高效液相色譜-串聯質譜法測定雞蛋中的氯黴素類獸藥殘留
-
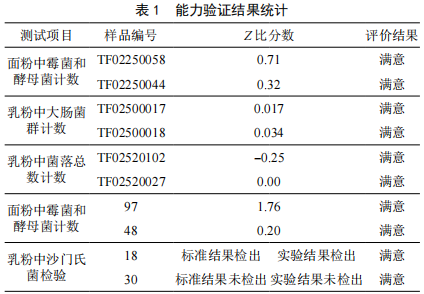
食品微生物檢驗實驗室質量控製——以陝西嘉禾藥業有限公司為例
-
安溪縣一起青褶傘中毒事件的調查
-

哈市市場監管局執法“巡回講堂”走進哈新區
-

利用色譜方法鑒定葡萄酒中摻假組分
-
高校反食品浪費方式的探析——以廣州新華學院為例
-

食品農藥殘留檢測中樣品前處理技術探究
-

烘烤對麵包質量的影響
-

杏仁胡蘿卜酥性餅幹的工藝研究
-
左旋薄荷酰胺的提純工藝研究
-

濕法消解-原子熒光法測定海產品總砷方法的改進
-

木瓜蛋白酶處理對牛肉品質影響的研究
-
超聲波輔助提取山核桃油工藝的研究
-
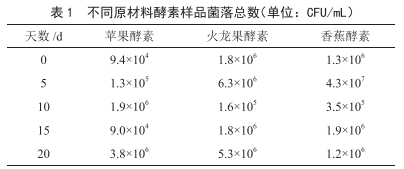
家庭自製酵素中的微生物汙染檢測
-

凱裏酸湯-白酸湯產品檢測及標準製定
-
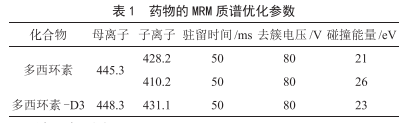
肉豬體內組織和體液中多西環素含量的相關性研究
-
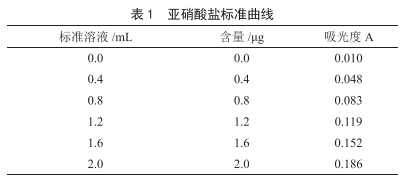
一起因食用韭菜餅引起亞硝酸鹽食物中毒原因的實驗室分析
-
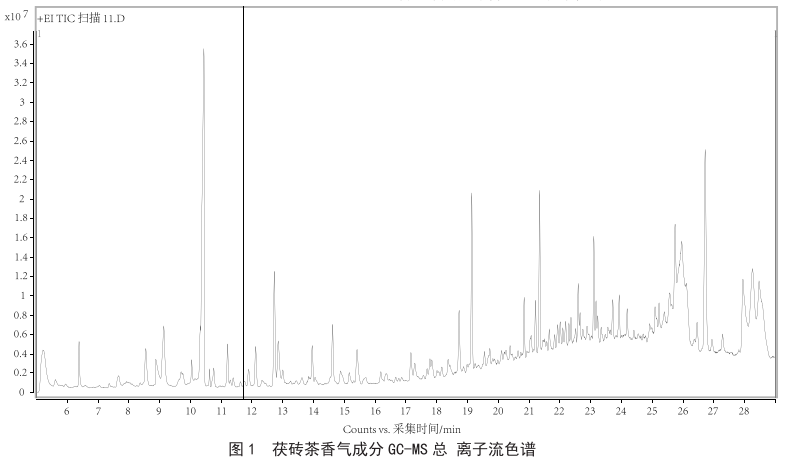
茯磚茶中特征揮發成分分析
-

GC-MS/MS法測定芹菜中毒死蜱殘留量 不確定度評定
-
免疫親和柱-高效液相色譜法測定餅幹中赭曲黴毒素A的含量
-

國際FAPAS乳粉中金黃色葡萄球菌定量檢測能力驗證結果與分析