氣相色譜-質譜法測定韭菜中5種農藥前處理方法的比較
2021-08-23 15:59:24 來源: 世界杯賽程預測 導刊
趙阿璿,李雪雪,槐歲絨,楊香娟
(鹹陽市食品藥品檢驗檢測中心,陝西鹹陽 712000)
摘 要:本文對韭菜中5種農藥(腐黴利、氟蟲腈、毒死蜱、氯氰菊酯、氯氟氰菊酯)殘留量進行了固相萃取和QuEChERS兩種前處理方法比較。結果表明,兩種淨化方法的5種農藥在0.01~0.50 µg/mL範圍內與目標物的響應值呈良好的線性關係,相關係數均大於0.9995,方法定量限均為0.01 mg/kg。采用QuEChERS法前處理時,5種農藥的平均回收率為83.0%~116.2%,為0.8%~4.4%;采用SPE法前處理時,5種農藥的平均回收率在80.0%~108.2%,為0.8%~4.6%。兩種前處理方法均滿足農藥殘留分析要求。(鹹陽市食品藥品檢驗檢測中心,陝西鹹陽 712000)
關鍵詞:韭菜;氣相色譜-質譜法;固相萃取;QuEChERS
為提高農作物產量,防治病蟲害,高毒性農藥的使用量越來越多。食品中特別是水果、蔬菜的農藥汙染通過食物鏈在人體積累,給人體健康帶來危害,但由於農藥殘留對人體產生的生理變化不明顯,往往被忽視[1]。目前農藥殘留所采用的檢測方法主要有氣相色譜法、氣相色譜-質譜聯用法、液相色譜法、液相色譜-質譜聯用法。由於韭菜基質較為複雜,天然色素含量較高,有揮發性含硫組分,尤其冷凍後的韭菜較難通過微波加熱消除巰基,因此使用上述方法檢測農藥殘留時,經提取淨化後方可上機[2]。
檢測農藥殘留常用的淨化方式有固相萃取、硫酸磺化、凝膠淨化、QuEChERS等[3-4],本文利用固相萃取和QuEChERS兩種淨化方式對比了韭菜中腐黴利、氟蟲腈、毒死蜱、氯氰菊酯和氯氟氰菊酯這5種農藥的殘留量,並進行了回收率、標準曲線、準確度、精密度等特性參數實驗,為相關工作者提供選擇。
1 材料與方法
1.1 主要儀器
氣相色譜質譜聯用儀,島津GCMS-TQ8040NCI,EI電子轟擊源;電子分析天平(梅特勒ME204);渦旋混合器(IKA VXR B S025);水浴恒溫振蕩器;高速離心機(湘儀);全自動濃縮儀(LabTech MV5);旋轉蒸發儀(廣東IKA RV-10)。
1.2 試藥
農藥的標準品:腐黴利、氟蟲腈、毒死蜱、氯氰菊酯、氯氟氰菊酯,以上標準溶液濃度均為1 000 µg/mL;環氧七氯B,濃度為100 µg/mL;試劑:氯化鈉(分析純,天津科密歐);乙腈(色譜純,TEDIA);乙酸乙酯(色譜純,TEDIA);甲苯(色譜純,TEDIA);QuEChERS淨化管:885 mg硫酸鎂、150 mgPSA、15 mgGCB;固相萃取柱:Cleanert S C18500 mg/6 mL,Agela;石墨碳黑-複合氨基複合柱 500 mg/500 mg/6 mL,Agela。
1.3 氣相色譜-串聯質譜分析條件
1.3.1 色譜條件
色譜柱為Rxi-5Mil MS石英毛線管色譜柱(30 m×0.25 mm×0.25 µm);載氣:氦氣,純度≥99.999%,流速1.69 mL/min;進樣溫度為220 ℃;進樣方式:不分流進樣,進樣體積1 µL;柱溫:初始溫度60 ℃,保持1 min,40 ℃/min升到120 ℃,保持0 min,5 ℃/min升到310 ℃,保持1 min。
1.3.2 質譜條件
離子源溫度為250 ℃,接口溫度為280 ℃,溶劑延遲13 min,多反應監測(MRM)采集模式,其他質譜條件見表1。

在市場上隨機采購10批次韭菜,將其全部切碎混勻,用四分法取樣,搗碎均質成漿,稱取樣品量1 kg放入聚乙烯瓶中,-18 ℃下保存。準確稱取10.0 g樣品,置於100 mL離心管中,加入20 mL乙腈,高速勻漿2 min,超聲振蕩提取30 min,加入4 g氯化鈉,高速勻漿1 min,以5 000 r/min離心5 min。
1.5 淨化方法
1.5.1 QuEChERS淨化法
準確吸取10 mL上清液於裝有900 mg無水硫酸鎂+150 mgPSA+15 mg GCB的15 mL塑料離心管中,渦旋混勻約1 min,5 000 r/min離心5 min。準確吸取淨化後的上清液5 mL到10 mL比色管中,40 ℃水浴中氮吹至近幹。加入20 µL的環氧七氯內標溶液,1 mL乙酸乙酯複溶,過0.22 µm有機微孔濾膜,用於測定。
1.5.2 SPE淨化法
將C18柱放入固定架上,加樣前先用10 mL乙腈預洗柱,下接雞心瓶,準確吸取10 mL上清液,並用15 mL乙腈洗滌柱,將收集的提取液和洗滌液在40 ℃水浴中旋轉濃縮至約1 mL,備用。用5 mL乙腈-甲苯(3+1)預淋洗石墨化碳黑-氨基複合固相萃取柱,棄去流出液。加入濃縮液,雞心瓶用2 mL乙腈-甲苯(3+1)分次淋洗,重複3次,將洗滌液轉移至柱中,再用25 mL乙腈-甲苯(3+1)洗脫,收集流出液,40 ℃水浴中旋轉濃縮至近幹。用正己烷定容至2 mL,加入內標液80 µL,混勻上機。
1.6 樣品的定性與定量分析
取經過前處理的樣品與農藥混合標準工作液1.0 µL,在氣相色譜/質譜條件下進行測定,保留時間及碎片離子可以進行定性、定量分析,用內標法計算樣品中的農藥濃度。檢測時,色譜峰保留時間在±5%內,在質譜圖中所選擇的離子對都出現,離子對豐度比和相近質量濃度標準品的離子豐度比一致,則該樣品中存在這種農藥。為減少基質的影響,定量時用陰性基質樣品配製混合標準工作溶液,標準溶液的濃度與待測化合物的濃度相近,要求曲線相關係數大於0.995。
2 結果與分析
2.1 方法的線性方程
分別按照QuEChERS淨化與SPE淨化方法製備兩種不同的陰性韭菜基質溶液,配製成質量濃度為0.01~0.50 µg/mL的標準工作液,基質標準溶液內標法定量。按照1.3的條件測定,結果見表2,兩種淨化方法的5種農藥在0.01~0.50 µg/mL範圍內與目標物的響應值呈良好的線性關係,相關係數均大於0.999,方法定量限均為0.01 mg/kg。
2.2 加標回收率和精密度
按照QuEChERS淨化與SPE淨化對陰性韭菜樣品進行加標回收試驗,選取高、中、低3個不同含量,6次平行測定,計算回收率、平均回收率、相對標準偏差。由表3知,采用QuEChERS法前處理時,5種農藥的平均回收率為83.0%~116.2%,為0.8%~4.4%;采用SPE法前處理時,5種農藥的平均回收率在80.0%~108.2%,為0.8%~4.6%。QuEChERS和SPE回收率比較結果表明,兩種淨化方式法測定同一種農藥同一個含量回收率存在差異,但均滿足農藥殘留分析要求。
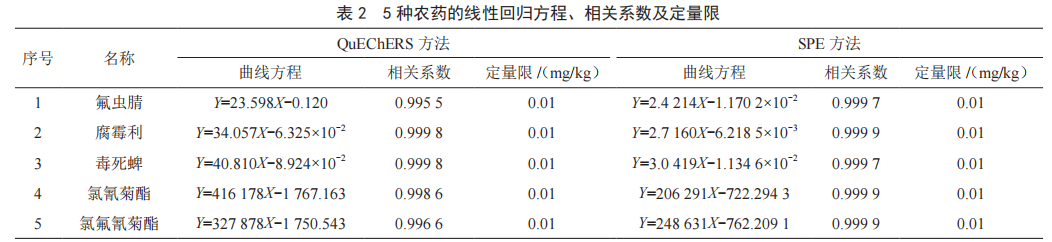

通過對兩種前處理方法的比較,發現兩種方法存在差異,但兩種前處理方法的定量限、回收率、精密度都滿足農藥殘留分析要求,可應用於實際樣品檢測。
參考文獻
[1]吳玉傑.食品中農藥殘留分析方法分析[D].長沙:湖南師範大學,2004.
[2]周勇,樸秀英,廖先駿,等.韭菜中腐黴利的殘留檢測及長期膳食暴露評估[J].農藥農學報,2021,23(2):373-379.
[3]國家衛生和計劃生育委員會,農業部,國家食品藥品監督管理總局.世界杯賽程預測國家標準 水果和蔬菜中500種農藥及相關化學品殘留量的測定氣相色譜-質譜法GB 23200.8—2016[S].北京:中國標準出版社,2016.
[4]國家衛生健康委員會,農業農村部,國家市場監督管理總局.世界杯賽程預測國家標準 植物源性食品中208種農藥及其代謝物殘留量的測定 氣相色譜-質譜聯用法:GB 23200.113—2018[S].北京:中國標準出版社,2018.






熱點推薦
-
特醫食品不是藥,卻是臨床治療的關鍵支撐
- 世界杯2022赛程时间表最新
-

全十紅紅稗餅幹|中秋團圓,回家必備
-

和汪氏蜂蜜共同來普及蜂蜜結晶現象
-

拿坡海開啟西餐加盟新潮流, 大眾化家庭小西餐成未來新趨勢
-
氣相色譜-質譜法測定韭菜中5種農藥前處理方法的比較
-
高效液相色譜法同時測定複雜食品基質中8種合成色素的含量
-

基於平衡積分卡的績效等級評價體係
-

利用超靈敏真菌毒素快檢法建立高效的食品加工過程質量監控體係
-
淺析食品中檢驗樣品的製備和管理
-

食品微生物檢驗菌落總數測定中不同檢測方法的應用分析
-
牛乳中脂肪酶活力檢測方法與影響因素研究
-

果蔬中農藥殘留快速檢測的質量控製探討
-

氣相色譜-質譜聯用法測定綠茶中4種菊酯類殺蟲劑農藥殘留量
-

幾種常見食品中亞硝酸鹽含量的測定
-
微波消解-電感耦合等離子體質譜法測定大米中的砷、鉛、鎘、鉻
-
2019—2020年北海市禽蛋中6種獸藥殘留檢測結果分析
-
食品中大腸菌群檢測結果與分析
-
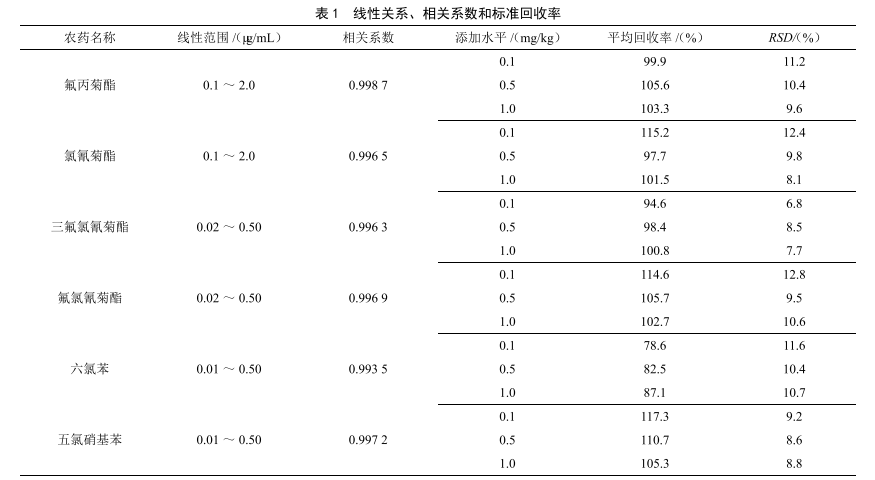
氣相色譜法測定大米中多種農藥殘留量
-
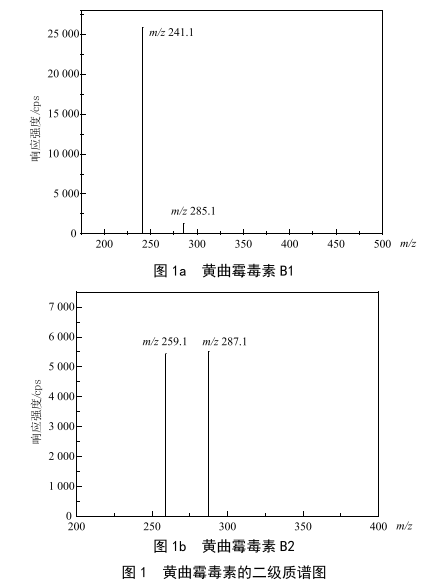
分散液液微萃取-超高效液相色譜-串聯質譜法測定茶飲料中黃曲黴毒
-

辣椒及其製品中辣椒素含量檢測及辣度分級
-
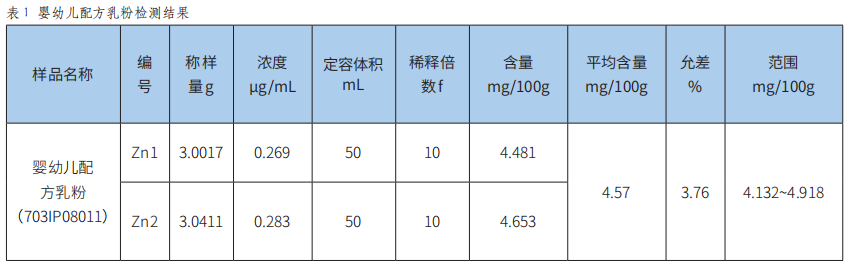
火焰原子吸收光譜法測定食品中鋅的方法驗證
-
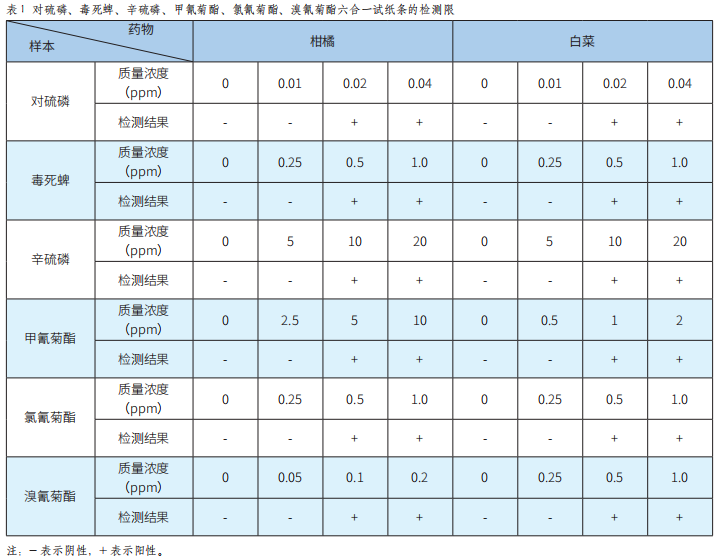
對硫磷等6種農藥殘留熒光免疫檢測芯片的研發及應用
-

山東省濰坊市市售蔬菜中農藥殘留現況及慢性膳食暴露風險評估
-

食品檢驗檢測機構質量控製的探討
-
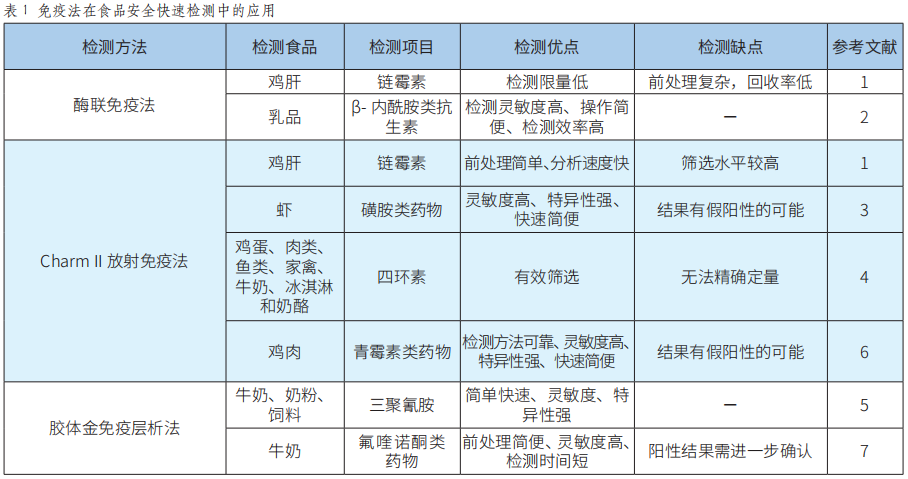
快速檢測技術在世界杯賽程預測 中的應用研究進展
-
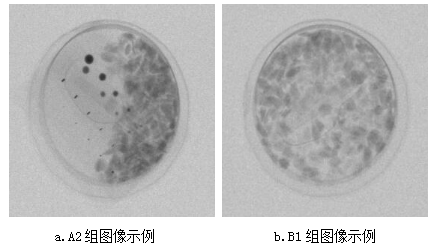
基於X射線圖像的堅果盒異物檢測
-
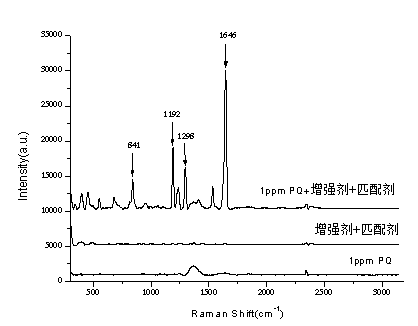
表麵增強拉曼光譜法快速檢測調味品中的百草枯
-
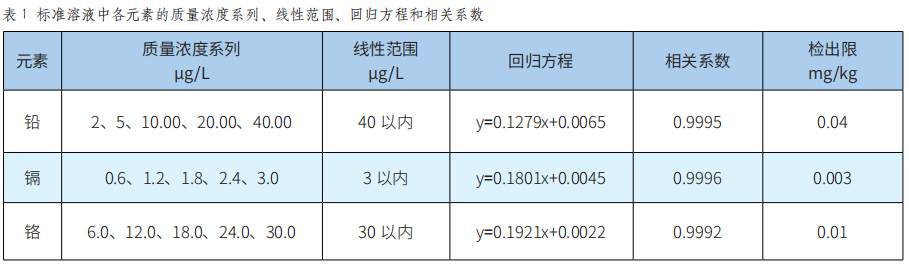
潞黨參重金屬汙染物含量的評估
-
電位滴定法測定特殊醫學用途食品中氯化物的不確定度評定