分散液液微萃取-超高效液相色譜-串聯質譜法測定茶飲料中黃曲黴毒
2021-08-04 15:42:51 來源: 世界杯賽程預測 導刊
分散液液微萃取-超高效液相色譜-串聯質譜法測定茶飲料中黃曲黴毒素B1和B2
陳佩佩1,2,陳琳珊1,2,陳 茹1,3
(1.廣東省食品工業公共實驗室,廣東廣州 511442;2.廣東省食品工業研究所有限公司,廣東廣州 511442;
3.廣東省質量監督食品檢驗站,廣東廣州 511442)
摘 要:建立了分散液液微萃取(DLLME)-超高效液相色譜-串聯質譜法(UPLC-MS/MS)對茶飲料中的黃曲黴毒素B1和黃曲黴毒素B2進行同時測定的方法。樣品以鹽酸為分散劑,3-氯苯胺為萃取劑進行萃取,調節pH至6後,離心分層,棄去上層水相,下層乙腈定容,使用電噴霧電離源正離子模式進行測定。結果表明,兩種黃曲黴毒素在1.0~100 μg/L範圍內呈良好線性關係,相關係數大於0.999 3,平均回收率為71.2%~91.0%,檢出限均為20.0 ng/kg,定量限均為50.0 ng/kg,相對標準偏差為2.23%~6.43%。該方法操作快速簡便、回收率好、靈敏度高、重現性好,適用於日常測定茶飲料中的黃曲黴毒素。
關鍵詞:分散液液微萃取;超高效液相色譜-串聯質譜;茶飲料;黃曲黴毒素
黃曲黴毒素(Aflatoxin,AFT)是由黃曲黴和寄生曲黴等某些菌株產生的一類次生代謝物[1],從化學結構上,均屬於雙呋喃香豆素衍生物,是一類毒性極強的雙呋喃環類毒素。由於AFT產生菌在自然界中廣泛存在,很多的動植物源性食品都有可能存在汙染[2],尤其在植物的種植、加工、運輸、儲存等過程都有可能被產生菌汙染,從而產生毒素[3]。同時,黃曲黴毒素在自然條件下穩定性較強,極性較低,難以用水或紫外線完全除去,隻有在強堿性條件下容易分解[4],過多的攝入引起致癌、致畸和致突變等毒性效應[5],其中以黃曲黴毒素B1的毒性致癌性最強。近年來,我國黃曲黴毒素汙染事件頻繁發生,因此,建立高效、通用、靈敏的相關檢測方法,對企業生產,消費者安全都有十分重要的意義。
目前,國內外對黃曲黴毒素的檢測方法主要有高效液相色譜法[6-7]、液相色譜-串聯質譜法[8-9]或液相色譜-高分辨質譜法[10]、免疫學檢測法[11-12]。高效液相色譜法操作煩瑣,盡管檢測器靈敏度高,但雜質多,分析時間較長,免疫學檢測法雖然能大批量檢測,但易與類似結構的化合物發生反應,出現假陽性,結果偏差較大。質譜法應用較廣,前處理簡單,定性定量能力好,但高分辨質譜價格昂貴,難以廣泛應用。本文以分散液液微萃取為前處理手段,建立了液相色譜-串聯質譜法對茶飲料中的黃曲黴毒素B1和B2進行測定。方法學驗證結果表明,方法整體方便快捷,靈敏度高,回收率好,可日常大批量檢測。
1 材料與方法
1.1 儀器與設備
API4000Q質譜儀、島津LC-20AD液相色譜係統、4k-15離心機、IKA Vortex4渦旋混勻器、Milli-Q Advantage A10 超純水係統。
1.2 材料與試劑
茶飲料,廣州市售;黃曲黴毒素B1、黃曲黴毒素B2,純度大於98%,德國Dr.Ehrenstorfer公司;甲酸、乙腈、3-氯苯胺、三氯甲烷,均為HPLC級,美國Thermo Fisher公司;鹽酸、磷酸鈉,均為分析純,廣州試劑廠;實驗用水為Milli-Q超純水。
1.3 樣品前處理
於15 mL尖底離心管中加入1 mL 2 mol/L的鹽酸溶液和50 μL 3-氯苯胺充分混勻,然後加入5 mL樣品,渦旋提取10 min,使用1 mol/L的磷酸鈉溶液調節pH至6,渦旋混勻後置於離心機中以4 500 r/min離心5 min,使3-氯苯胺沉澱。除去上層水相,下層3-氯苯胺用乙腈定容至
200 μL,待測定。
1.4 標準曲線的繪製
標準儲備溶液:分別準確稱取黃曲黴毒素B1和黃曲黴毒素B2標準品各0.050 0 g於100 mL棕色具塞容量瓶中,用乙腈稀釋成500 mg/L的儲備液。
標準中間溶液:用乙腈將上述儲備液稀釋成濃度為
10 mg/L的標準中間液。
溶劑曲線:使用乙腈配製成1.0 µg/L、2.0 µg/L、5.0 µg/L、10 µg/L、20 µg/L、50 µg/L和100 µg/L的標準使用液,繪製校準曲線。
基質曲線:使用空白烏龍茶基質液配製校準曲線,濃度點與溶劑曲線相同。
1.5 色譜及質譜條件
1.5.1 色譜條件
色譜柱:Xbridge BEH C18色譜柱(100 mm×2.1 mm,
3.0 µm);進樣體積:10 µL;流速0.5 mL/min;柱溫:35 ℃;流動相為乙腈(A)-0.1%甲酸(B);等度洗脫程序:0.0~
5.0 min,75% A。
1.5.2 質譜條件
離子源:ESI源;離子化模式:電噴霧電離源正模式(ESI+);氣簾氣壓:241 kPa;毛細管電壓:5 500 V;離子源溫度(TEM):550 ℃;霧化氣壓力:345 kPa;加熱輔助氣壓力:345 kPa;碰撞氣(CAD):中等;采集模式:多反應監測(MRM)模式。
2 結果與分析
2.1 定性定量分析依據的收集
根據黃曲黴毒素B1和黃曲黴毒素B2的化學結構,其含有較多含O基團,在離子化過程中傾向於得到質子,因此分別以其相對分子質量加上H+質量,初步確定母離子質荷比。以混合流動相的流動注射的方式向離子源注射標準溶液,在/ 313.1和/ 315.1處,均得到較高響應的[M+H]+離子,同時以多反應監測(MRM)模式采集二級碎片。進一步優化去簇電壓DP和碰撞電壓CE,得到較優響應,二級質譜圖見圖1,詳細質譜信息見表1。
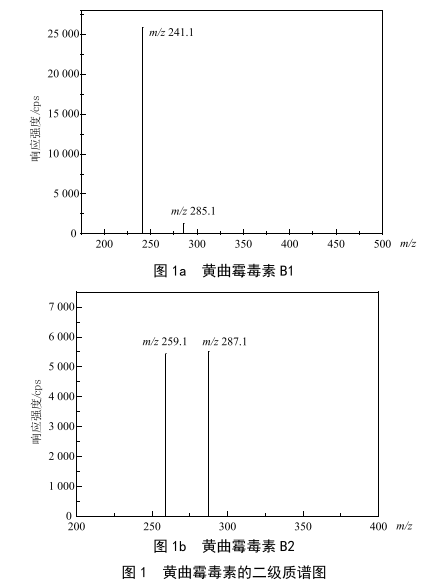
2.2 分散劑的選擇
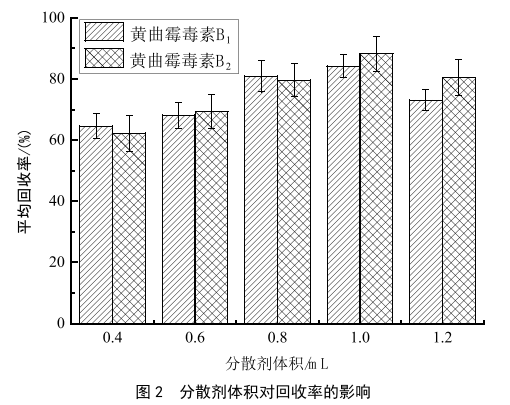
分散液液微萃取過程中,加入分散劑和待測樣液互溶,加入萃取劑振蕩形成微小的液滴,這些液滴在移動的過程中於對目標物進行連續萃取,形成水/分散劑/萃取劑形成均相乳濁液體係,快速完成分析物在水溶液與萃取劑之間的分配平衡。選擇鹽酸作為分散劑。在本實驗中,當分散劑用量小於0.6 mL時,提取回收率低於70%,使用量為1.0 mL時,回收率達到最高,隨著使用量提高至1.2 mL,回收率有所回降。所以鹽酸分散劑用量選取為1.0 mL。詳細結果見
2.3 萃取劑體積的選擇
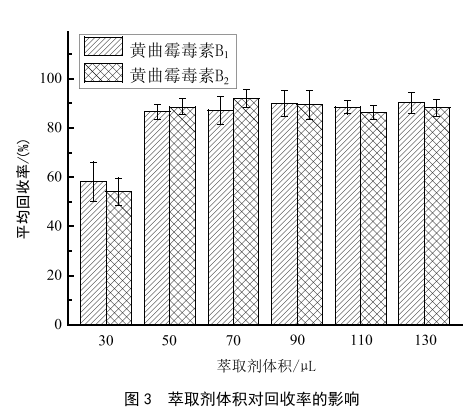
在分散劑體積為1 mL的條件下,分別用30 μL、
50 μL、70 μL、90 μL、110 μL和130 μL的3-氯苯胺進行萃取,考察了不同體積的萃取劑對萃取回收率的影響,結果如圖3所示。隨著3-氯苯胺體積增大,回收率也增大,而當體積大於50 μL時,回收率增大不明顯,而且3-氯苯胺的使用體積越大,pH回調的幅度也越大,實驗更耗時,最終定容液3-氯苯胺的占比也越大,對於含有甲酸的流動相體係,容易造成溶劑效應,使峰型變差。因此,最終確定3-氯苯胺50 μL為最佳萃取體積。
2.4 基質效應的探討
市售的茶飲料會添加糖、色素、酸度調節劑或其他添加劑,且含量遠超於黃曲黴毒素可能存在的含量,加上茶葉本身含有較多的天然物,因此有可能造成較嚴重的基質效應(ME),使定量結果偏差較大。為了探究實驗實際存在的基質效應,分別用空白基質液和乙腈配製了基質曲線和純溶劑曲線,並以基質曲線和純溶劑曲線的斜率比值為判斷依據,當ME在80%~120%時,則說明基質效應不明顯。實驗結果顯示,黃曲黴毒素B1和黃曲黴毒素B2的值分別為23.2%和26.5%,說明基質負效應明顯,因此最終選擇使用基質曲線進行定量,以校正基質效應造成的偏差。
2.5 定量分析結果
使用空白基質溶液配製標準曲線,線性範圍為1.0~100 µg/L,將該曲線於優化後的條件下測定,以質量濃度(X)為橫坐標,峰麵積(Y)為縱坐標,繪製標準曲線。同時以信噪比/=3和信噪比/=10分別確定方法的檢測限與定量限。黃曲黴毒素B1和黃曲黴毒素B2定量離子流圖見圖4。
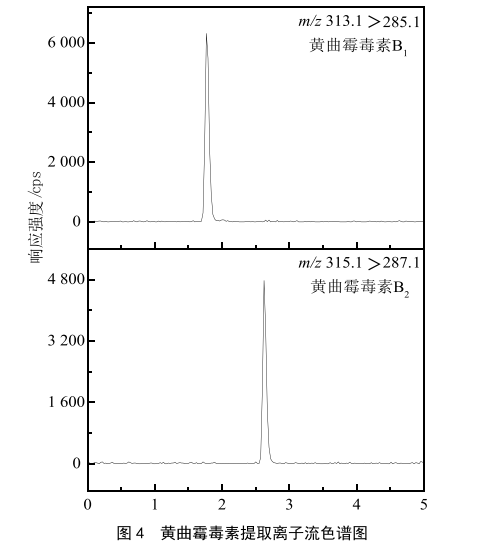
結果表明,黃曲黴毒素B1和黃曲黴毒素B2的相關係數分別為0.999 6和0.999 3,基質曲線方程分別為=5 050+
2 240和=3 090+2 750,檢測限均為20.0 ng/kg,定量限均為50.0 ng/kg。由分析結果可知,本方法定量能力好,檢出限低,可有效對茶飲料中的黃曲黴毒素進行定量分析。
2.6 回收率及精密度分析
分別對市售的3種茶飲料(綠茶、烏龍茶、紅茶)進行測定,結果均為陰性。使用該樣品進行加標回收實驗。每種樣品均按照定量限的1倍、2倍和10倍進行添加,即0.05 μg/kg、
0.10 μg/kg、0.50 μg/kg濃度水平,每個濃度水平在日內平均測定6次,平均回收率和相對標準偏差結果見表2。
3種樣品的平均回收率範圍在71.2%~91.0%,範圍在2.23%~6.43%,方法回收率、精密度及準確度良好,可用於實際樣品的測定。

3 結論
以液液微萃取為基礎,建立前處理方法,方法整體高效簡單,通過調節pH,較大限度回收萃取劑,提高了回收率;建立UPLC-MS/MS法,優化了色譜及質譜條件,兩種黃曲黴毒素在樣3 min內出峰,且峰型良好。使用基質曲線對定量結果進行校準,降低了基質效應對結果的影響。回收率和相對標準偏差數據表明,方法靈敏度高、重現性好,可為茶飲料中毒素測定方法的開發提供
依據。
參考文獻
[1]潘中華,徐燕芳,成恒嵩.黃曲黴毒素分析方法進展[J].農業環境與發展,1995,12(2):30-33.
[2]趙亞榮,劉香香,趙曉麗,等.生薑及其製品中常見真菌毒素汙染與檢測方法研究進展[J].農產品質量與安全,2020(4):49-54.
[3]NAIR K P P.The Postharvest and industrial processing of ginger [M].Oxford: Elsevier,2013.
[4]楊惠宇,趙瑩,潘發林.魚類飼料中黃曲黴毒素B1檢測技術研究進展[J].河北漁業,2020(8):52-55.
[5]OMOTAYO O P, OMOTAYO A O, BABALOLA O O, et al. Dataset on the toxic effects of aflatoxin and ochratoxin A on the human gastric smooth muscle cells[J].Data in Brief, 2019,25:104089.
[6]華麗霞,曾華蘭,蔣秋平,等.免疫親和淨化-光化學衍生液相色譜檢測不同樣品中的黃曲黴毒素[J].中國農業科技導報,2020,22(7):181-187.
[7]石格鑫.高效液相色譜法測定小麥中黃曲黴毒素B1含量[J].現代食品,2020(10):204-206.
[8]孫夏榮,葛曉明,王建花.QuEChERS超高效液相色譜-串聯質譜法檢測中藥飲片中黃曲黴毒素[J].中國藥業,2020,29(11):44-47.
[9]華宇,高和楊,聶興娜,等.同位素內標-高效液相色譜-串聯質譜法檢測牛奶及奶粉中黃曲黴毒素M1[J].世界杯賽程預測質量檢測學報,2020,11(6):1978-1984.
[10]姚婷,王丹,李雙,等.超高效液相色譜-四極杆-飛行時間質譜法快速檢測發酵黑茶中黃曲黴毒素B1[J].分析測試學報,2017,36(11):1346-1351.
[11]Iqbal J, Asghar M A, Ahmed A, et al. Aflatoxins contamination in Pakistani brown rice: a comparison of TLC, HPLC, LC-MS/MS and ELISA techniques[J]. Toxicol Mech Method, 2014,24(8):544-551.
[12]Juki H, Dedi S, Rodi M, et al. Determination of Aflatoxin M1 in Raw Milk by the ELISA Method in the Una-Sana Canton[M]. 30th Scientific-Experts Conference of Agriculture and Food Industry,2020.
作者簡介:陳佩佩(1990—),女,廣東茂名人,大專,助理工程師。研究方向:食品分析檢測。






熱點推薦
-

全十紅紅稗餅幹|中秋團圓,回家必備
-

和汪氏蜂蜜共同來普及蜂蜜結晶現象
-

拿坡海開啟西餐加盟新潮流, 大眾化家庭小西餐成未來新趨勢
-

摩購空間“霸王餐”來襲 打破傳統營銷套路帶動商家業績翻倍
-

開學季食安隱患猛增 禧雲食安開展公益行動助力意識提升
-
分散液液微萃取-超高效液相色譜-串聯質譜法測定茶飲料中黃曲黴毒
-

辣椒及其製品中辣椒素含量檢測及辣度分級
-
火焰原子吸收光譜法測定食品中鋅的方法驗證
-
對硫磷等6種農藥殘留熒光免疫檢測芯片的研發及應用
-

山東省濰坊市市售蔬菜中農藥殘留現況及慢性膳食暴露風險評估
-

食品檢驗檢測機構質量控製的探討
-
快速檢測技術在世界杯賽程預測 中的應用研究進展
-
基於X射線圖像的堅果盒異物檢測
-
表麵增強拉曼光譜法快速檢測調味品中的百草枯
-
潞黨參重金屬汙染物含量的評估
-
電位滴定法測定特殊醫學用途食品中氯化物的不確定度評定
-
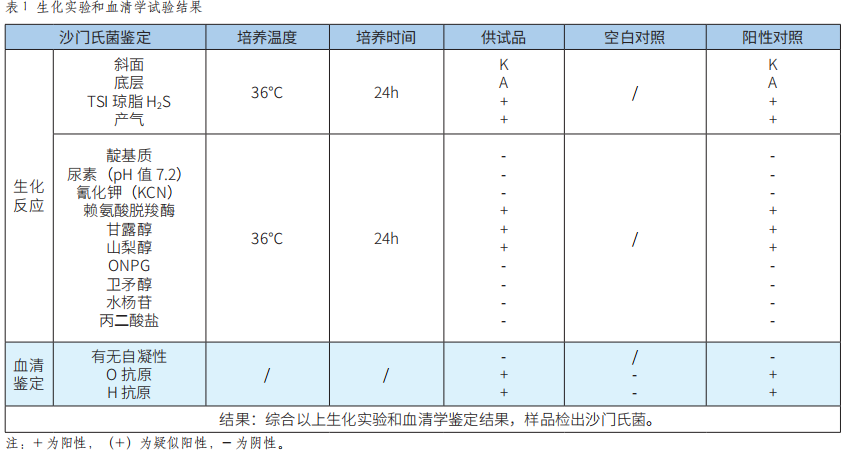
API 20E鑒定係統等兩種檢測方法應用於食品中沙門氏菌的檢測結果
-

2020年上海市青浦區肉製品中單增李斯特菌分子分型及耐藥性分析
-
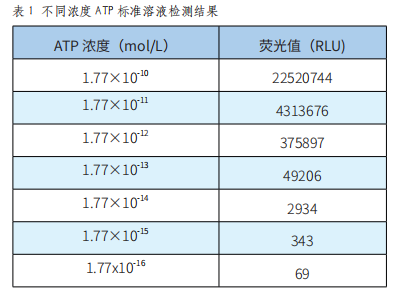
一種基於ATP熒光反應的潔淨度檢測係統的開發與驗證
-
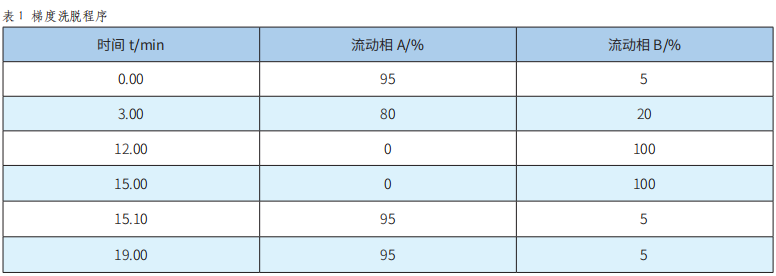
固相萃取-高效液相色譜-二極管陣列檢測法測定飲料中9種人工合成著
-

一種檢測鹽黴素膠體金試紙條的研製及其應用
-

高效液相色譜法測定綠豆糕中5種常見的食品添加劑
-
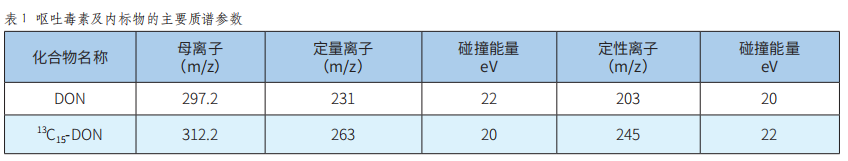
液相色譜-串聯質譜內標法測定全麥粉中嘔吐毒素含量的不確定度評
-
淺析QuEChERS方法結合色譜串聯質譜技術在果蔬農藥殘留檢測中的優勢
-
食品檢測技術問題及其解決措施研究
-
梅特勒-托利多最新版 ProdX™ 軟件引入工業 4.0 功能
-
氣相色譜-質譜法測定曼陀羅藥酒中莨菪堿的含量
-

食品中雞源性成分標準檢測方法的比較性研究
-
基於銀沉積的微間隙陣列電極檢測鹽酸克倫特羅的方法研究
-
2019年榆林市監督抽檢餜餡與糖琪子的酸價、過氧化值檢測結果分析