高效液相色譜檢測水產品中甲氧苄啶殘留量
2019-07-26 13:55:54 來源: 世界杯賽程預測 導刊
摘要:本文建立了水產品中甲氧苄啶殘留量的高效液相色譜(HPLC)的檢測方法。該方法操作簡便,準確度高,適用於水產品中甲氧苄啶殘留檢測。
關鍵詞:高效液相色譜;水產品;甲氧苄啶
目前,水產品中甲氧苄啶的檢測方法主要有紫外分光光度法[4-5]、吸附溶出伏安法[6]、液相色譜法等[7-9]。本文以市售的鮮魚為研究對象,對其中的甲氧苄啶檢測前處理條件進行了選擇和優化,高效液相色譜紫外檢測器檢測條件的建立和優化,建立了高效液相色譜法檢測水產品中甲氧苄啶的方法。
結果與討論
樣品前處理條件的優化
(1)提取溶劑的選擇。水產品基質複雜,且含有較多的脂肪和蛋白等成分,而甲氧苄啶屬於弱堿性化合物,易溶於酸性溶液,而純水相的提取液在淨化時易出現乳化,不利於分層,因此本研究中選擇酸化的甲醇作為提取液。考慮樣品基質中含有蛋白,硫酸具有一定的沉澱蛋白的作用,因此選擇硫酸化的甲醇溶液,分別考察了甲醇-硫酸體積比為7:3、6:4、5:5、4:6、3:7時對水產品中甲氧苄啶的提取效果,當比例為7:3時效果最好,因此最終選擇甲醇-硫酸體積比7:3的溶液作為提取液。
(2)淨化條件的優化。在酸性條件下,甲氧苄啶幾乎不溶於三氯甲烷,而水產品中含有較多的脂肪、蛋白和部分色素等,三氯甲烷可以除去脂。分別考察了在提取過程中和提取之後加入三氯甲烷的淨化和回收效果,發現在提取過程中加入三氯甲烷,首次提取液未見溶入三氯甲烷,但在第二次提取過程中,有部分三氯甲烷溶於提取液中,提取液體積明顯大於理論加入體積,最終造成提取液中溶入了三氯甲烷相,引入雜質,淨化效果變差,同時因雜質的引入,使得回收率不到70.0%;與此同時我們在提取過程之後向合並後的提取液中加入三氯甲烷,未發現有三氯甲烷相溶於提取液中的情況,且回收率均在80.0%以上,因此選擇在提取過程之後加入三氯甲烷進行淨化。
(3)線性方程與定量限。按照1.4項的方法配製6水平標準曲線,並在優化色譜條件下進行檢測,以甲氧苄啶的峰麵積為縱坐標,甲氧苄啶的濃度為橫坐標繪製曲線,甲氧苄啶的線性範圍為0.05~1.0 μg/mL,相關係數r為0.9998,線性關係良好,定量限為15 μg/kg。
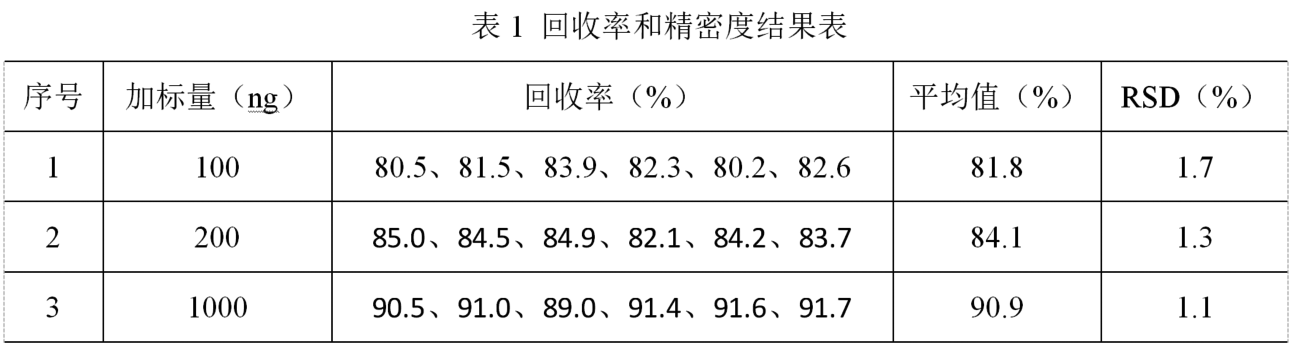
(4)回收率和精密度。在空白樣品中添加甲氧苄啶標準溶液,製備含量分別為20、40和200 μg/kg的加標樣品,按照1.4項處理方法及儀器測定條件進行測試,每個添加水平平行測定6份,結果見表1。各化合物的平均回收率為80.2~91.7%,相對標準偏差為1.1~1.7%,采用本方法檢測水產品中的甲氧苄啶含量,準確度和精密度均較好。
實際樣品檢測
采用本研究建立的檢測方法,對市售的10批次鮮魚進行檢測,其中3批檢出甲氧苄啶,但檢出值均小於農業部235號公告附錄2:已批準的動物性食品中最高殘留限量規定的限量要求50 g/kg範圍內,可以放心食用。
結論
本研究采用酸化的甲醇提取水產品中甲氧苄啶,用三氯甲烷除脂等雜質,二氯甲烷反萃取的方法,結果準確可靠,精密度高,重現性好。與國家標準《GB 29702-2013水產品中甲氧苄啶殘留量的測定》相比,操作簡單、快速,除雜效果更優,適用於食品檢驗檢測機構進行水產品中的甲氧苄啶的檢測。
佟曉波 孫曉娟 張京華
遼寧省食品檢驗檢測院






熱點推薦
-

2019《世界杯賽程預測 導刊》雜誌訂閱返百元紅包!
-

世界杯賽程預測 謠言治理的法律分析
-

又一大省連續發生兩起非洲豬瘟疫情!
-
9月大事 | 市場監管總局開展2018年全國“質量月”活動
-

世界杯賽程預測 ,我們怕的是什麼?
-
高效液相色譜檢測水產品中甲氧苄啶殘留量
-

液相串聯質譜法測定蔬菜中三氯殺蟎醇
-
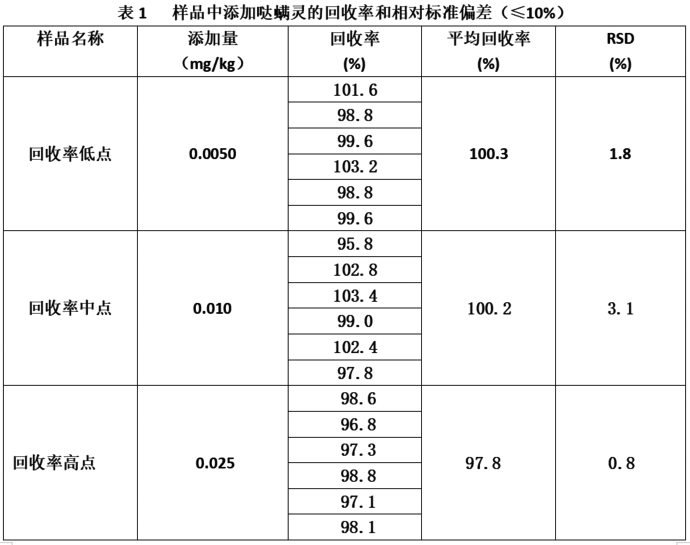
氣質聯用法測定茶葉中噠蟎靈殘留量
-
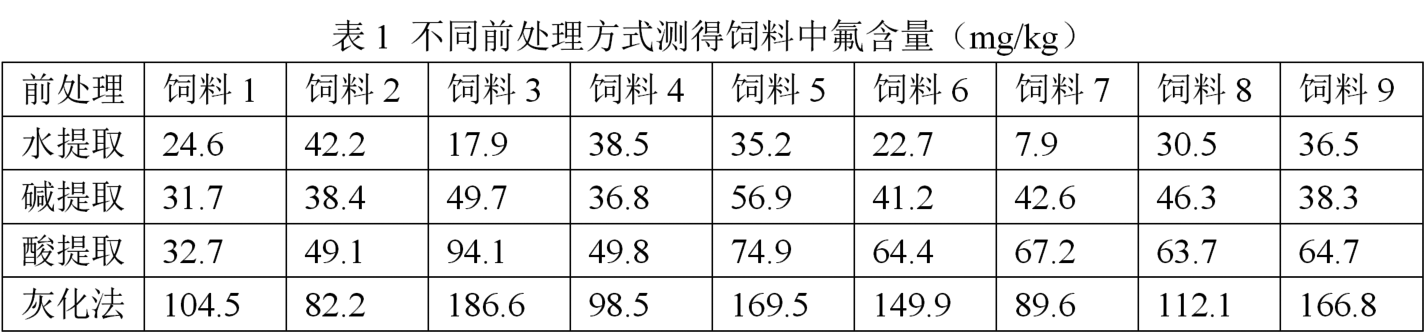
氟離子選擇電極法測定飼料中氟的方法研究
-

薑黃素抗氧化的初步機理研究
-

瓜爾膠在食品中的應用
-

保健食品你有在吃嗎?
-

食想,動物蛋白的未來
-

美國雜誌刊登新研究發現 現在的食品都太甜了
-

飲用純淨水≠飲用天然水≠飲用天然礦泉水
-

減少脂肪和糖類吸收——ID-alG™褐藻提取物
-

食品中的甲醛從哪來
-

高糖分飲食難逃患癌風險
-

用“互聯網+”守護舌尖安全
-

飲料世界杯賽程預測 控製要做到這些
-

植物提取物標準促行業規範發展
-

Science子刊:這種常用的食品添加劑,吃太多真的會變胖!
-

依案說法——淺論食用農產品的定性及標簽規定
-

想要世界杯賽程預測 ?先做個有知識的吃貨
-
奶粉中維生素B2標準物質的研製
-

高效液相色譜和超高效液相色譜法測定乳製品中三聚氰胺含量的對比
-
南瓜魔芋果凍的工藝配方研究
-
液相質譜測定中離子加合應用的一些研究
-

火爆全網的吐司先生,分分鍾就能饞哭你!
-

收藏這種方法,一鍵解決農殘最多的5樣菜